Research lines
How does a single genome sequence encode the information to build the enormous complexity of cell types and structures of an adult animal? How are changes in this sequence translated into morphological differences during evolution? These two exciting questions have always been the center of my research. In my lab, we approach these topics focusing on cell and tissue type specific transcriptomes: How are they encoded in the genome? How are they generated during embryogenesis? How do they impact cell function in adult organisms? How do they evolve and how they impact evolution? What are their pathological implications? To answer these questions, we not only study transcriptional regulation, but also other mechanisms that expand transcriptomic diversity, such as alternative splicing and gene duplication, combining computational and experimental approaches using in vitro and in vivo systems.
Functional impact of microexons in neurons and sensory cells

Background: Mirroring their unparalleled morphological and cellular complexity, vertebrate nervous systems harbor the highest levels of cell type specific alternative splicing in nature. Among them, there are hundreds of tiny exons, or microexons (2014). Microexons are switched on during neuronal differentiation by their master regulators Srrrm3 and Srrm4, modulate protein-protein interactions, and are mis-regulated in autism spectrum disorder (2016, 2018). Not surprisingly, they are highly conserved across vertebrates; in fact, microexons and their regulators originated in the ancestors of bilaterians (2019), and a similar molecular program is present in fruit flies (2022). Recently, we discovered a set of microexons specifically expressed in the retina essential for photoreceptor development and vision (2022).
Open questions: How do Srrm3/4 promote microexon inclusion in different neuronal subtypes? How do microexons impact protein function? What are their roles in high-order functions such as social behavior or sleep? Are mutations around microexons relevant for retinal diseases? Are there microexons specific to other sensory neurons?
Approaches: Single-cell long-read sequencing, patient WGS, biochemical and parallel splicing minigene assays, behavioral experiments in mutant mouse and zebrafish models, brain and retina organoids.
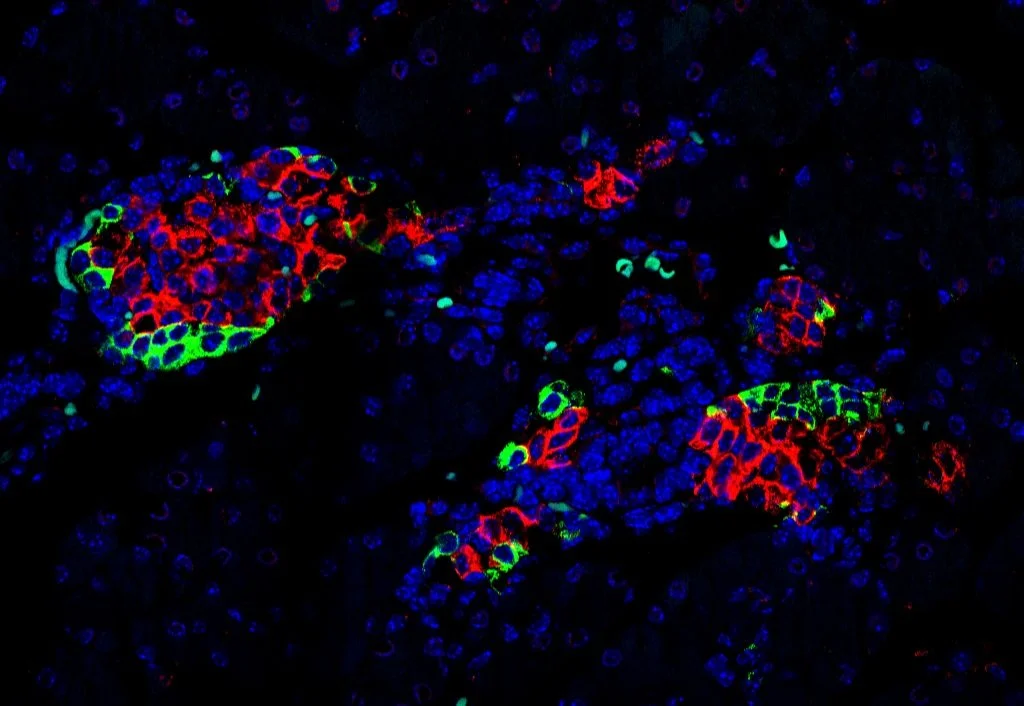
Functional impact of microexons in endocrine pancreas

Background: Diabetes is a metabolic disorder associated with severe clinical complications resulting from dysfunction or loss of insulin-secreting pancreatic beta cells. While the study of transcriptional alterations has received much attention, little is known about the importance of alternative splicing for this disease and for beta cell function. Remarkably, a subset of neuronal microexons, highly sensitive to the action of their master regulator SRRM3, is also expressed in endocrine pancreatic cells (IsletMICs, 2023). IsletMICs are enriched in genes involved in vesicle transport, exocytosis and linked to type 2 diabetes risk, and their mis-regulation leads to alterations in insulin secretion and glucose homeostasis. We also found human genomic variants in SRRM3 linked to glycemia and diabetes risk.
Open questions: What are the cis-regulatory basis of the different microexon sensitivities to their master regulator? How do IsletMICs impact metabolism and insulin exocytosis in beta cells? What is their role in endocrine pancreas development and evolution? Can IsletMICs be used as therapeutic targets in diabetes?
Approaches: Single-cell long-read sequencing, biochemical and parallel splicing minigene assays, imaging, physiological tests in conditional KO mouse models, islet organoids.
Assembly and evolution of cell-type specific gene modules

Background: Cell types depend on sets of interconnected gene regulatory networks and gene modules for their specification and unique functional features. Therefore, to know how cell types originate and evolve new properties, we need to understand how gene modules are acquired and integrated during evolution. We have previously studied the stepwise de novo assembly of various tissue-specific exon networks regulated by key master regulators: Nova (2011, 2021), Esrp (2017) and Srrm3/4 (2019, 2022). Now, we are interested in how individual genes/exons or whole gene/exon modules get integrated into pre-existing cell type networks to provide new cellular features or even to give rise to novel cell types in evolution.
Open questions: How has gene module co-option contributed to the origin of new vertebrate cell types, such as the electrocytes of electric fish or sensory neuron subtypes? Are genes co-opted by enhancer re-purposing or through new enhancers? How do new individual genes/exons get integrated into pre-existing networks?
Approaches: single-cell multi-omics, single-cell long-read transcriptomics, spatial transcriptomics, in situ hybridization and IHC, reporter transgenesis.
Evolution of tissue-specific transcriptomes across bilaterians
Background: Animals are formed by tens of distinct cell types. These are all generated from a single set of genes, usually expressed across multiple cell types. This raises a major paradox: how can evolution optimize the function of a gene for a given cell type without jeopardizing its function in other cell types? Luckily, evolution has ways to specialize cell type proteomes: gene duplication and alternative splicing. By assembling a vast RNA-seq dataset for eight tissues across twenty bilaterians, we study how animals deploy these specialization mechanisms. For instance, nearly half of the ancestral bilaterian genes have evolved tissue-specific expression, usually through gene duplication and subsequent expression specialization of one of the copies (2023). Importantly, this mechanism has been particularly relevant after the two vertebrate whole genome duplications.
Open questions: What is the differential contribution of gene duplication and alternative splicing to tissue-specific specialization in vertebrates and in insects? What genes have evolved tissue-specificity in bilaterians and what are their phenotypic implications? How evolutionarily conserved are these mechanisms?
Approaches: Comparative transcriptomics and genomics, gene/exon orthology inferences, experimental evolution in model systems.
Evolution of zygotic genome activation across metazoans

Background: One of the few universal developmental processes across animals is the maternal-to-zygotic transition, in which embryogenesis goes from being orchestrated by the maternal transcripts and proteins deposited in the oocyte to being controlled by active transcription of the zygotic genome. Zygotic genome activation (ZGA) is thus a fundamental step for subsequent cell differentiation and organogenesis. However, the stage at which the ZGA occurs, and the transcriptomic changes associated with it, remain a mystery for most animal lineages. This applies to newly transcribed protein-coding genes, but also to transposable elements and splice isoforms. In that regard, we showed that a programmed splicing failure occurs at ZGA in mammals, contributing to modulate DNA damage responses (2022). To investigate ZGA across metazoans, we generated time-course RNA-seq for 10 animal phyla.
Open questions: What developmental and genomic features contribute to the timing of ZGA? What genes are activated during ZGA? What transposable elements are more often activated at ZGA? Are programmed splicing failures a general feature of ZGA?
Approaches: stage-specific RNA-seq, comparative transcriptomics and genomics.
Evolution of microsynteny and CNEs across vertebrates

Background: Genome regulation relies on an intricate 3D organization. Cis-regulatory elements can be located hundreds of kbps away from their target gene promoter, intermingled within other genes; however, enhancers must be in physical contact with promoters to exert their function. Consequently, disruption of TADs can have pathological implications and evolutionary consequences. These regulatory constraints in turn preserve gene order, or microsynteny, forcing pairs of genes to be together for millions of years. In fact, studies of microsynteny conservation, including ours (2012, 2012, 2020), provided some of the first indications of the importance of the 3D organization of animal genomes. Behind this ancient regulatory conservation are also the conserved non-coding elements (CNEs), regulatory sequences strictly preserved across vertebrates. Our current hypothesis is that CNEs act as hubs for multi-gene contacts, contributing to conserve microsynteny.
Open questions: Are vertebrate CNEs more often involved in multi-gene enhancer-promoter contacts? How often are new contacts established and disrupted across vertebrate evolution? What are the functional implications of CNE-promoter contact turnover?
Approaches: capture HiC, ATAC-seq, comparative genomics, CRISPRi/CRISPRa in neural progenitors.
Evolutionary Medical Genomics (EvoMG)

Background: Evolutionary Medical Genomics (EvoMG) aims at applying evolutionary principles to better understand the genomic basis of disease and improve human health. Research in EvoMG covers a wide range of topics, including variant effect prediction from phylogenetic analysis, cancer evolution, evolutionary history of disease, viral evolution, microbiomes, and antibiotic resistance. The CRG and UPF have launched a Joint Program on EvoMG, coordinated by our lab. Within this framework, and taking advantage of our knowhow and resources on both evolutionary and medical transcriptomics, we are engaging on several EvoMG projects. We will harness RNA-seq data from a wide range of tissues and species to investigate transcriptomic susceptibilities to disease and to identify clinically relevant alterations.
Open questions: Can we model transcriptomic variation across tissues and species to identify clinically relevant transcriptomic alterations? Are there evolutionary differences in background susceptibilities to cancer among species? What can pancreas adaptations to different lifestyles tell us about diabetes risk? How conserved is cell type specific aging among vertebrate retinae?
Approaches: bulk RNA-seq, single-cell long-read sequencing, mathematical modeling, comparative transcriptomics.